

What is a Biological Design Tool?
Summary of State of the Art Biological AI (Part I in two part series)


A picture from Ernst Haeckel Kunstformen der Nature
by Hans Gundlach
What is a Biological Design Tool? Biological Design Tools usually refer to AI models specifically trained on biological data and intended for biological tasks for example protein structure or sequence predictions. These contrast with broader AIs like GPT that are trained on a wide range of tasks. However, one of the points in this essay is that there isn’t a strong distinction between biological design tools and other AIs like GPT. I want to give a basic sense of how some of these models work. This includes how they make predictions, how they are used, and how they are trained.
Background: I’m assuming some familiarity with recent developments in machine learning. If not ask your grandson what he thinks about Yann LeCun. I have some added sections that go into more technical detail for those interested. I’ll also assume some knowledge of biology. For example, do you know what protein residue is? If not watch this video. If you have neither you're probably cool anyway just smile and nod.

What I usually think about when I hear the words '“biological design tools” ( I actually got this for my 11th birthday).
What Do Biological Models Do?
Most models that are classified as biological take in sequence ie protein sequence, genome, etc, and predict some important properties of it. For protein sequences, this includes 3D- structure, protein-ligand interactions, finding binding sites, solubility, stability, etc while for genetic sequences this includes things like finding promotors, splice sites, etc. There are also protein design models which take a sequence of required properties (ie required structures, solubility, stability) and convert these to a protein sequence that can be developed. There are also general-purpose biological models that predict almost all of these properties for whatever sequence you are interested in. Given that nature depends on so many different types of sequences like DNA, RNA, and protein. It is not surprising that ….
Most BDT Models are Sequence Models:
Below is a list of BDT tools. Out of all these models, RFdiffusion is the only one that is not primarily based on sequence prediction. These models are fed a biological sequence like TAT and are asked to predict the next letter A. You can also see that most of these models are instances of transformers (The same AI architecture as Chat GPT). Why transformers? Transformers can make sequence predictions that pay attention to sequence elements far back in a sequence. For example, regulatory elements such as enhancers of a given gene can exist up to 100,000 base pairs from the promotor (transcription start site). AI architectures that only pay attention to small regions will miss these relationships. This includes convolution architectures like the original AlphaFold. However, Transformers also require compute resources which scale as the square of the sequence length. For genetic sequences, this is a crucial disadvantage which is why newer AI architectures like MAMBA and Hyena which scale more modestly outperform transformer models on DNA sequence tasks.
Here’s a Sample of State of the Art BDT tools:
a transformer-based model that uses sequence and protein structure data to make predictions.
similar design to AlphaFold2. ESMfold trains a protein sequence model and then feeds this attention data into a folding module.
protein folding language model
transformer-based protein design model. This model converts 3d geometric information from a desired protein shape to a sequence which is fed into the model.
DNA sequence model based on Hyena-AI-architecture which shares similarities with convolution architectures.
A selection state model that performs much better than Transformers for long sequences. It has shown impressive abilities in DNA sequence prediction.
transformer model for protein-ligand interactions
transformer model for general-purpose biological data ie predicts folds, interactions, binding sites, etc. This is the largest model of the ones I’ve mentioned and is the only one that would be required to report its training based on the new AI executive order.
Diffusion-based protein folding model.
Why Does Predicting the Next _____ in a Sequence Work So Well?
How is it that a model that is only built to predict the next element in a DNA or Protein Sequence can understand the three-dimensional structure of a protein? Well, to make sequence predictions you have to understand life. There are repeat elements like the Alu element that has a million copies in the human genome. Yet there are also more subtle relationships. For example, you can discover protein binding sites by looking at how models pay attention to specific protein sequences. The sequence surrounding a given binding site will be heavily influenced by the binding site. All the protein elements are coordinated and selected in the perfect way to bind to a given ligand (for example iron in hemoglobin). As an analogy, if you pay attention to the phrase “Uber for …” you can easily predict what I’m going to say next.
In addition, learning which protein elements are in contact can help an AI model predict the next element of a protein sequence. For example, if you say “Grandson” and the date is close to now you are likely to hear “birthday” in the same sentence. if you “hear” Cystein and the sequence looks like Insulin you should be on the lookout for another Cystein because proteins use two Cystein’s to form disulfide bonds when they fold.
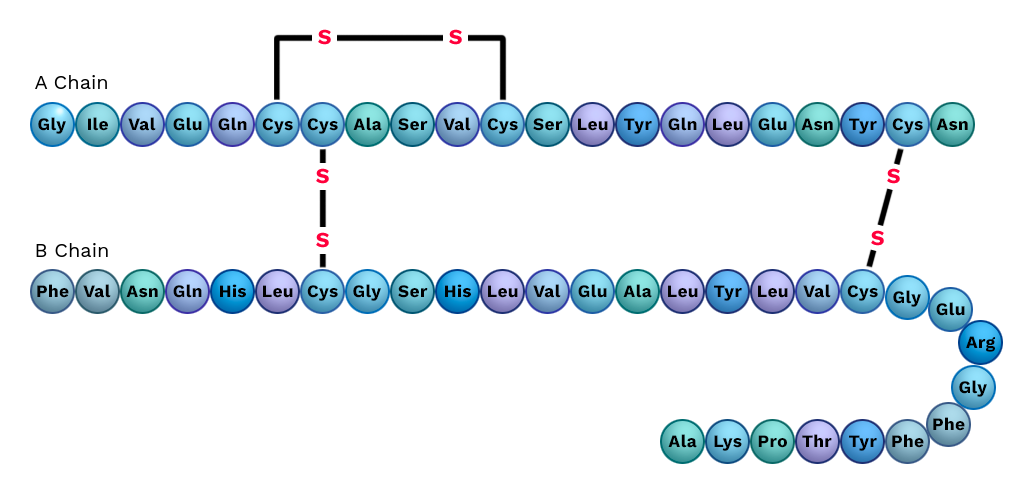
Example of Disulfide bonds in insulin after cutting (taken from Labster Theory)
{kind=link}
MSA Data:
Another way to see how models infer structure from sequence data is to look at what is called MSA or multi-sequence alignment. MSA is not used in all protein folding or biological models. However, it illustrates the intimate connection between sequence data and biology. MSA information includes sequences that are related to the sequence given to the AI. For instance, you can have protein sequence data from a similar protein in a related animal (ie orthologous) or sequences from similar proteins in the same organism (paralogs). There’s a lot you can infer from this data. If two amino acids seem to mutate in pairs then it probably means they are in contact. If two amino acids are in contact ie bonding in some form then changing just one would lead to loss of the bond, protein function, loss of protein use, death, and loss of such sequence in the protein library. Therefore, if a pair of residues seem to have correlated changes then you can infer they are in contact or do something important together.
A visual illustration of how MSA can be used to infer contact from correlation
Taken from Improved contact prediction in proteins
(Warning Technical ML): Interpretability in Protein Language Models:
So what are these language models really learning about biology? Are these models using this sequence data to actually fold these proteins or just to pattern-match data?
To answer this question researchers looked at the performance of these models on protein isoforms. These are protein structures that have very similar sequences to a full-length protein yet are unfolded and nonfunctional. If these models are physically folding these proteins then they should be able to accurately predict these structures. However, language models fail quite badly on this task. In reality, these models are pattern-matching a lot of information about the paring of residues (see Zhang et al). This makes sense with what we discussed earlier. The model recognizes that it's reading a sequence similar to Insulin and learns basic pairings that are common ie Cystein-Cystein disulfide bond.
Nevertheless, looking at the internal structures in these models can give a lot of biological insight. Individual attention heads in protein transformer models can be used to infer things like protein element distances, binding sites, turns bends, helixs, sheets, etc. Some protein folding models like ESMFold simply use this attention information along with an external folding module to determine the fold of a given protein.

Visual depiction of how protein transformers pays specific attention to binding sites when making predictions. (this depicts one attention head of a protein sequence transformer taken from Vig et al.)
What’s Next:
I hope this post gives an idea of the power of these models. The next post in the sequence (pun intended) outlines what sort of responsibility comes with this power.
 The X-ray … | Flickr")
*This can now be done on NVIDIA GPU